
Reaktionsmechanismen und Katalysatordesign
Das wesentliche Forschungsziel in der Gruppe „Katalysatoren von Kohlenwasserstoffen“ ist, Beiträge zur Entwicklung erneuerbarer Quellen von Kohlenstoff für den Energieverbrauch der Gesellschaft zu liefern. Durch Modellierung mit periodischer Dichtefunktionaltheorie (DFT) und ab initio Berechnungen an cluster Modellen erlangen wir ein Verständnis solcher Prozesse auf atomarer Ebene. Die wichtigsten Programmpakete, die wir verwenden, sind VASP, TURBOMOLE und ORCA. Die hauptsächlichen Themen in der Gruppe sind die Umsetzung von Methanol zu Kohlenwasserstoffen, die gezielte Funktionalisierung von Biomasse und die Untersuchung von Katalysatordeaktivierung.
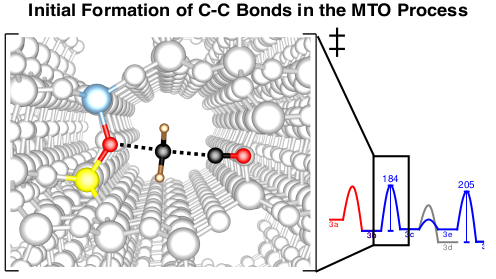
Synthese von Kohlenwasserstoffen aus Methanol
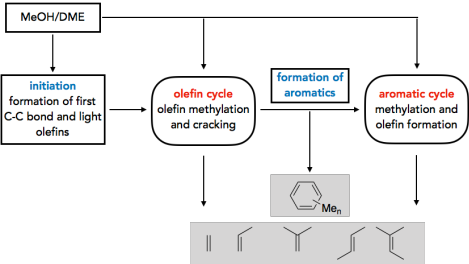
Es gibt ein großes Interesse an Prozessen, die Methanol selektiv in Produkte wie Kohlenwasserstoffe, Alkene und Kraftstoffe umwandeln. Das Verständnis der Reaktionsmechanismen kann hierbei zur Verbesserung von Selektivitäten und Ausbeuten der verwendeten Katalysatoren beitragen. Eine Frage von besonderer Bedeutung ist die Initiierung des methanol-to-olefins (MTO) Prozesses. Während es allgemein anerkannt ist, dass die Reaktivität im stationären Zustand durch katalytisch aktive Kohlenwasserstoffe bestimmt wird, die den sogenannten‚ hydrocarbon pool’ (HCP) bilden, ist weniger gut erforscht, wie sich dieser HCP bildet. Wir untersuchen dieses Problem durch die Berechnung vieler möglicher Reaktionen, wobei wir hochgenaue CCSD(T) und MP2-Rechnungen mit periodischen DFT-Rechnungen kombinieren1-3. Kinetische Modellierungen zeigen, dass die direkte C-C Bindungsbildung durch Methanol tatsächlich einen realistischen Mechanismus dargestellt, wobei der günstigste Mechanismus über die Bildung von CO und dessen weitere Methylierung abläuft.4 Neben der Initiierung untersuchen wir auch die Rolle der Olefine5 und Aromaten6 im HCP.
Alternative Ansätze zu Erzeugung von Kraftstoffen aus erneuerbaren Quellen
Ein anderer interessanter Prozess ist die Synthese von Oxymethylendimethylethern (OMEs) aus Methanol und Formaldehyd. OMEs haben großes Potential als Zusatzstoffe für Diesel gezeigt, aber die Selektivität für OMEs mit der gewünschten Kettenlänge bleibt eine Herausforderung. Typischerweise werden saure Katalysatoren, wie etwa Zeolithe verwendet. Wir beschäftigen uns hauptsächlich mit der Reaktion von Trioxan (TOX) mit OME1 als alternative Edukte. Die direkte Insertion von TOX ist hierbei energetisch günstiger als die schrittweise Zerlegung zu Formaldehyd, was die experimentell beobachtete, anfängliche Selektivität für OME4 erklärt7.
Metall-Oxid Wechselwirkung und Katalysatordeaktivierung
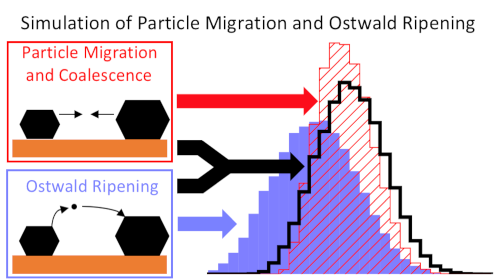
Ein weiterer Forschungsschwerpunkt der Gruppe ist Katalysatordeaktivierung, insbesondere durch Sinterung. Hier beschäftigen wir uns unter anderem mit der Entwicklung verbesserter Sinterungsmodelle5. Neben der klassischen, Oberflächen-vermittelten Ostwald Reifung sind wir vor allem an Gasphasen-vermittelter Reifung interessiert. Dieser Mechanismus kann für Platin relevant sein, wo molekulares PtO2 bei hohen Temperaturen und unter oxidierenden Bedingungen gebildet werden kann. Unsere Untersuchen mit mean-field Modellen zeigen, dass dieser Mechanismus tatsächlich realistisch ist und experimentelle Sinterungskinetiken erklären kann.9 Weiterhin untersuchen wir Metall-Oxid Wechselwirkung in Form von Adhesion und der Bildung von ‚single atoms‘.10-11
Publikationen
- Plessow, P. N.; Studt, F., Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations. ACS Catal. 2017, 7 (11), 7987-7994.
- Goncalves, T. J.; Plessow, P. N.; Studt, F., On the Accuracy of Density Functional Theory in Zeolite Catalysis. ChemCatChem 2019, 11 (17), 4368-4376.
- Plessow, P. N.; Studt, F., How Accurately Do Approximate Density Functionals Predict Trends in Acidic Zeolite Catalysis? J. Phys. Chem. Lett. 2020, 11 (11), 4305-4310.
- Plessow, P. N.; Smith, A.; Tischer, S.; Studt, F., Identification of the Reaction Sequence of the MTO Initiation Mechanism Using Ab Initio-Based Kinetics. J. Am. Chem. Soc. 2019, 141 (14), 5908-5915.
- Plessow, P. N.; Studt, F., Olefin methylation and cracking reactions in H-SSZ-13 investigated with ab initio and DFT calculations. Catal. Sci. Technol. 2018, 8 (17), 4420-4429.
- Fečík, M.; Plessow, P. N.; Studt, F., A Systematic Study of Methylation from Benzene to Hexamethylbenzene in H-SSZ-13 Using Density Functional Theory and Ab Initio Calculations. ACS Catal. 2020, 10 (15), 8916-8925.
- Goncalves, T. J.; Arnold, U.; Plessow, P. N.; Studt, F., Theoretical Investigation of the Acid Catalyzed Formation of Oxymethylene Dimethyl Ethers from Trioxane and Dimethoxymethane. ACS Catal. 2017, 7 (5), 3615-3621.
- Dietze, E. M.; Abild-Pedersen, F.; Plessow, P. N., Comparison of Sintering by Particle Migration and Ripening through First-Principles-Based Simulations. J. Phys. Chem. C 2018, 122, 26563−26569.
- Plessow, P. N.; Abild-Pedersen, F., Sintering of Pt Nanoparticles via Volatile PtO2: Simulation and Comparison with Experiments. ACS Catal. 2016, 6 (10), 7098-7108.
- Goodman, E. D.; Johnston-Peck, A. C.; Dietze, E. M.; Wrasman, C. J.; Hoffman, A. S.; Abild-Pedersen, F.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Catalyst deactivation via decomposition into single atoms and the role of metal loading. Nat. Catal. 2019, 2 (9), 748-755.
- Dietze, E. M.; Plessow, P. N., Predicting the Strength of Metal–Support Interaction with Computational Descriptors for Adhesion Energies. J. Phys. Chem. C 2019, 123 (33), 20443-20450.