
Theoretische Katalyse

Der Übergang zu Lösungen im Bereich der erneuerbaren Energien erfordert einen starken Fokus auf die Katalyse, da die Umwandlung von einer chemischen Form in eine andere mit größtmöglicher Effizienz erfolgen muss. Die Arbeitsgruppe „Theorie und Modellsysteme“ arbeitet an dem tieferen Verständnis von Katalysatoren und erforscht wie diese auf atomarem Niveau funktionieren. Dieses Verständnis kann durch Quantenchemische Rechnungen erreicht werden. In der heterogenen Katalyse werden für diese Rechnungen aufgrund der Größe der Modellsysteme normalerweise auf der Dichtefunktionaltheorie (DFT) basierende Methoden eingesetzt. DFT Rechnungen können hier Parameter wie Adsorptionsenergien, Chemisorptionsenergien von Intermediaten auf katalytischen Oberflächen und Reaktionsbarrieren liefern. Durch Kopplung dieser Rechnungen mit mikrokinetischen Modellen von Oberflächenprozessen kann hierbei ein tieferes Verständnis der Faktoren die die katalytische Aktivität limitieren erlangt werden. Die Identifizierung von Schlüsselfaktoren die sowohl die Aktivität als auch die Selektivität eines Katalysators beschreiben kombiniert mit dem Wissen wie diese Faktoren sich hinsichtlich verschiedener Materialien verändern erlaubt die Konstruktion von sogenannten Aktivitätsvulkanen bei denen sich der optimale Katalysator durch eine moderate Bindung zu den Reaktionsintermediaten auszeichnet. Diese Strategie der Katalysatoranalyse ermöglicht das schnelle rechenbasierte Screening für neue katalytische Materialien und wurde durch die Entdeckung neuer Materialen mittels Rechnungen erfolgreich für Übergangsmetallbasierte Katalysatoren demonstriert. Bisher ist dieser Deskriptoren basierte Ansatz allerdings nur für Reaktionen mit relativ wenigen Intermediaten angewendet worden und unsere Gruppe entwickelt diese Methoden weiter um höhere Komplexität wie Adsorbat-Adsorbat Wechselwirkungen, Oberflächendiffusion und mannigfaltige Aktive Zentren zu berücksichtigen. Diese Komplexität steht im Zentrum der Betrachtung katalytischer Prozesse die mit der effizienten Umwandlung von Biomasse zu synthetischen Kraftstoffen verbunden sind.
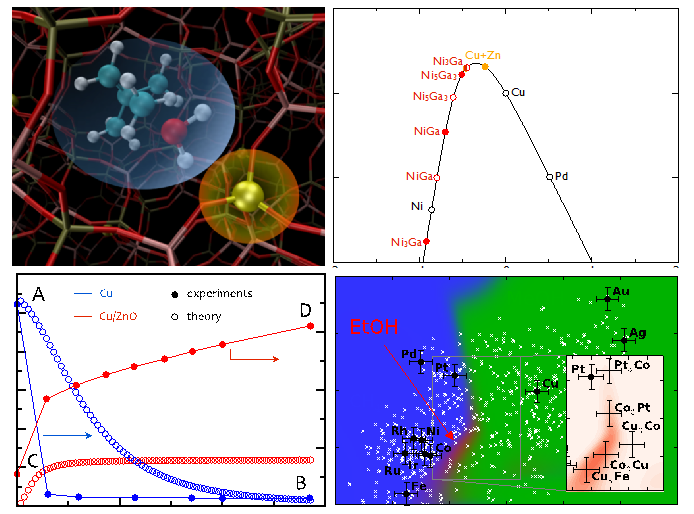
Übergangszustand der Buten-Methylierung. Aktivitätsvulkan der CO2 Hydrierung. Vergleich theor. und exp. kinetischer Daten der Methanol Synthese. Computergestütztes Screening nach Katalysatoren für die Synthese von höheren Alkoholen aus Synthesegas.
Des Weiteren beschäftigt sich unsere Gruppe auch mit der Säure-Base Katalyse, z.B. auf Zeolithen basierend, da eine große Anzahl von katalytischen Prozessen von dieser Art Katalyse abhängig ist. Die Entwicklung von Design Strategien für diese und andere Materialklassen steht noch ganz am Anfang. Große Ressourcen sind erforderlich um die Entwicklung so weit heranzutreiben, um das computergestützte Design dieser Materialien zu ermöglichen. Die Genauigkeit der theoretischen Methoden die als Grundlage für die Vorhersage von Katalysatoreigenschaften dienen ist auch ein kritisches Forschungsfeld. Unsere Gruppe hat deswegen auch Methoden für die Quantifizierung von Ungenauigkeiten in der Berechnung von Oberflächenprozessen im Fokus.