
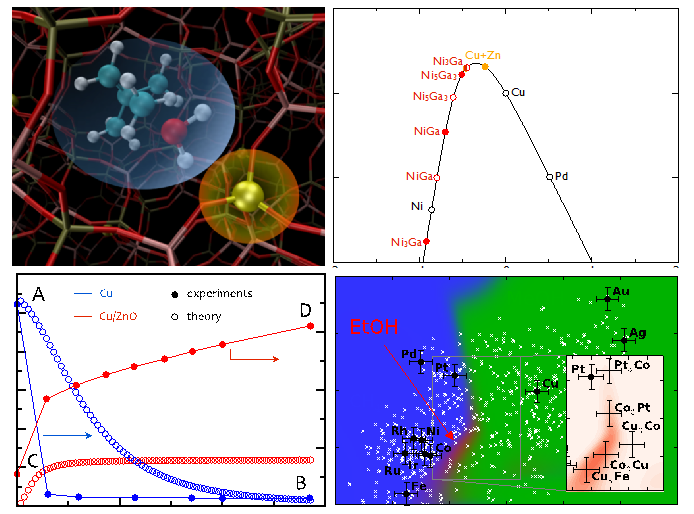
Theoretical catalysis

The drive for sustainable energy solutions requires a strong focus on catalysis, as it is essential to be able to efficiently transform energy from one chemical form into another. The research of the group “Theory and Model Systems” aims at an understanding of how catalysts work at the atomic-scale. This understanding can be obtained through quantum chemical calculations. In the field of heterogeneous catalysis, these are usually based on density functional theory (DFT). DFT calculations can be used to obtain parameters like adsorption energies, chemisorption energies of intermediates on catalytic surfaces and reaction barriers. If these calculations are coupled with (micro-)kinetic models of surface processes an in-depth understanding of the factors that limit the catalysts performance can be achieved. Identifying key descriptors that ultimately determine the activity and selectivity of materials and establishing those descriptors for a variety of catalysts allows for the construction of volcano-shaped curves where the optimal catalyst is characterized by an intermediate bond strength of the reaction intermediates. This strategy allows for the fast computational screening of new catalytic materials and has been successfully demonstrated for (transition-)metal based catalysts where the first examples of computationally discovered materials emerged. So far this descriptor-based approach has only been shown for reactions involving few intermediates and our group is developing this methodology further to include more complexity such as adsorbate-adsorbate interactions, surface diffusion, and multiple active site motifs. This complexity plays a central role for catalytic processes that can efficiently convert biomass to synthetic fuels.
We also target acid-base catalysis, based on e.g. zeolites, as a large number of catalytic processes will rely heavily on on these types of catalysts. The development of design strategies for these and other classes of catalysts is still in its infancy and large resources are required to develop approaches that enable the in-silico design of novel catalysts based on those materials. The accuracy of theoretical calculations that are the basis for theoretical predictions is also a critical issue. Our group hence devotes specific focus to methods that allow for the quantification of uncertainties in the predictions of surface processes using DFT calculations.