
Reaction mechanisms and catalyst design
Group mission
The primary research goal of the “Reaction Mechanisms and Catalyst Design” group is to contribute to the development of renewable sources of carbon for society’s energy needs. Through modeling using periodic density functional theory (DFT) and ab initio calculations on periodic and cluster models, we gain an understanding of such processes at the atomic level. The main software packages we use are VASP, TURBOMOLE, and ORCA. The primary research topics in the group are the conversion of methanol to hydrocarbons, the targeted functionalization of biomass, and the study of transition metal catalysts.
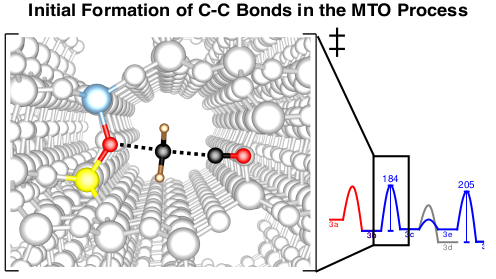
Synthesis of Hydrocarbons from Methanol
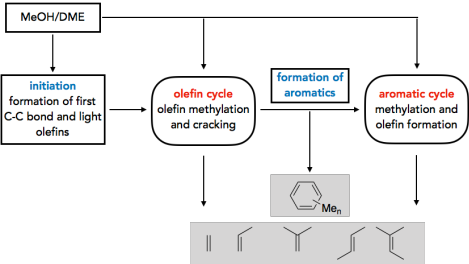
There is considerable interest in processes that selectively convert methanol into products such as hydrocarbons, alkenes, and fuels. Understanding the reaction mechanisms can help improve the selectivity and yield of the catalysts used. A question of particular importance is the initiation of the methanol-to-olefins (MTO) process. While it is generally accepted that reactivity in the steady state is determined by catalytically active hydrocarbons that form the so-called “hydrocarbon pool” (HCP), less is known about how this HCP forms. We investigate this problem by calculating many possible reactions, typically combining highly accurate CCSD(T) and MP2 calculations with periodic DFT calculations. 1–4 Kinetic modeling shows that direct C–C bond formation via methanol indeed represents a realistic mechanism, with the most favorable mechanism proceeding via the formation of CO and its subsequent methylation.5 In addition to initiation, we also investigate the role of olefins6, aromatics7-10, and other cyclic compounds11 in the HCP.
Methods for the Accurate Calculation of Reaction Mechanisms in Zeolites
Highly accurate ab initio methods, such as DLPNO-CCSD(T) in combination with complete basis set (CBS) extrapolated MP2 calculations, can only be performed for non-periodic cluster models. In practice, they can therefore only be applied to stationary points and are combined with the harmonic approximation for the motion of atomic nuclei. The dynamics of the nuclei can be simulated more accurately and generally using semiclassical Born-Oppenheimer molecular dynamics (MD) calculations. Intrinsic reaction barriers of adsorbed species can be calculated using techniques such as umbrella sampling or blue moon sampling. A central problem here is the adsorption and desorption of reactants and products, which is essential for an accurate description of catalytic processes. To this end, we have been involved in the development of various methods for calculating free energies of adsorption12–14. In combination with free energy perturbation theory (FEPT), free energies can now be determined using MD simulations, enabling predictions of reaction kinetics even for complex reaction mechanisms15.
Structure and Dynamics of Transition Metal Catalysts
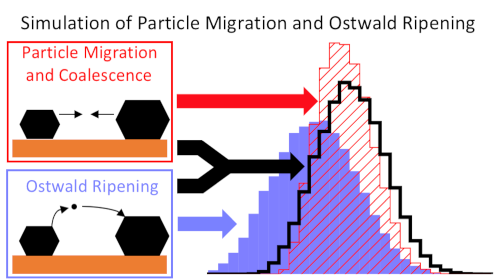
Theoretical studies often focus on calculating reaction mechanisms using models of active sites, such as the fcc(111) or fcc(211) surface. Our goal is to predict the structure and stability of the catalysts under reaction conditions. In addition to surface energies, the interaction with the support material (usually an oxide) is the most important factor determining the shape of the metal particles. The investigation of surfaces and interfaces using DFT, taking reaction conditions into account, provides their stability and allows the prediction of the equilibrium structure16–18. Furthermore, we investigate the deactivation of catalysts via sintering19–22. In addition to monometallic catalysts, we also study the properties of alloyed catalysts18, 23–25. In this context, we also use machine learning models that can predict not only stability but also the spectroscopic properties of adsorbates.15
Publications
1. Plessow, P. N.; Studt, F., Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations. ACS Catal. 2017, 7, 7987-7994.
2. Goncalves, T. J.; Plessow, P. N.; Studt, F., On the Accuracy of Density Functional Theory in Zeolite Catalysis. ChemCatChem 2019, 11, 4368-4376.
3. Huber, P.; Plessow, P. N., The role of decarboxylation reactions during the initiation of the methanol-to-olefins process. J. Catal. 2023, 428.
4. Huber, P.; Plessow, P. N., Accurate and Efficient Description of Acidic Zeolites with Plane-Wave Density Functional Theory Using Range-Separated Hybrid Functionals. ChemPhysChem 2025, e2500147.
5. Plessow, P. N.; Smith, A.; Tischer, S.; Studt, F., Identification of the Reaction Sequence of the MTO Initiation Mechanism Using Ab Initio-Based Kinetics. J. Am. Chem. Soc. 2019, 141, 5908-5915.
6. Plessow, P. N.; Studt, F., Olefin methylation and cracking reactions in H-SSZ-13 investigated with ab initio and DFT calculations. Catal. Sci. Technol. 2018, 8, 4420-4429.
7. Fečík, M.; Plessow, P. N.; Studt, F., A Systematic Study of Methylation from Benzene to Hexamethylbenzene in H-SSZ-13 Using Density Functional Theory and Ab Initio Calculations. ACS Catal. 2020, 10, 8916-8925.
8. Plessow, P. N.; Enss, A. E.; Huber, P.; Studt, F., A new mechanistic proposal for the aromatic cycle of the MTO process based on a computational investigation for H-SSZ-13. Catal. Sci. Technol. 2022, 12, 3516-3523.
9. Plessow, P. N.; Studt, F., Cooperative Effects of Active Sites in the MTO Process: A Computational Study of the Aromatic Cycle in H-SSZ-13. ACS Catal. 2022, 13, 624-632.
10. Enss, A. E.; Plessow, P. N.; Studt, F., Theoretical investigation of the paring mechanism of the MTO process in different zeolites. J. Catal. 2024, 432.
11. Vicente, H.; Huber, P.; Gayubo, A. G.; Studt, F.; Plessow, P. N., Reactivity of Pentamethylcyclopentenyl Cations toward Olefin Formation in the Methanol-to-Olefin (MTO) Process. J. Phys. Chem. C 2025, 129, 20971-20980.
12. Amsler, J.; Plessow, P. N.; Studt, F.; Bucko, T., Anharmonic Correction to Adsorption Free Energy from DFT-Based MD Using Thermodynamic Integration. J. Chem. Theory Comput. 2021, 17, 1155-1169.
13. Amsler, J.; Plessow, P. N.; Studt, F.; Bucko, T., Anharmonic Correction to Free Energy Barriers from DFT-Based Molecular Dynamics Using Constrained Thermodynamic Integration. J. Chem. Theory Comput. 2023, 19, 2455-2468.
14. Plessow, P. N., Anharmonic Adsorption Free Energies and Apparent Activation Barriers for Mobile Reactants Based on Molecular Dynamics Simulations. J. Chem. Theory Comput. 2025.
15. Vicente, H.; Plessow, P. N., Formaldehyde formation and its impact on the MTO initiation investigated with ab initio molecular dynamics simulations. J. Catal. 2026, 457.
16. Chen, J.; Sharapa, D.; Plessow, P. N., Stability and formation of hydroxylated α−Al2O3(0001) surfaces at high temperatures. Phys. Rev. Res. 2022, 4, 013232.
17. Plessow, P. N.; Campbell, C. T., Influence of Adhesion on the Chemical Potential of Supported Nanoparticles as Modeled with Spherical Caps. ACS Catal. 2022, 12, 2302-2308.
18. Hejral, U.; Plessow, P. N.; Franz, D.; Shipilin, M.; Gutowski, O.; Rütt, U.; Noei, H.; Vonk, V.; Stierle, A., Composition-Dependent Alloy Nanoparticle Shape Changes under Reaction Conditions: Kinetic and Thermodynamic Effects. J. Phys. Chem. C 2024, 128, 4330-4342.
19. Plessow, P. N.; Abild-Pedersen, F., Sintering of Pt Nanoparticles via Volatile PtO2: Simulation and Comparison with Experiments. ACS Catal. 2016, 6, 7098-7108.
20. Goodman, E. D.; Johnston-Peck, A. C.; Dietze, E. M.; Wrasman, C. J.; Hoffman, A. S.; Abild-Pedersen, F.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Catalyst deactivation via decomposition into single atoms and the role of metal loading. Nat. Catal. 2019, 2, 748-755.
21. Goodman, E. D.; Carlson, E. Z.; Dietze, E. M.; Tahsini, N.; Johnson, A.; Aitbekova, A.; Nguyen Taylor, T.; Plessow, P. N.; Cargnello, M., Size-controlled nanocrystals reveal spatial dependence and severity of nanoparticle coalescence and Ostwald ripening in sintering phenomena. Nanoscale 2021, 13, 930-938.
22. Aitbekova, A.; Zhou, C.; Stone, M. L.; Lezama-Pacheco, J. S.; Yang, A. C.; Hoffman, A. S.; Goodman, E. D.; Huber, P.; Stebbins, J. F.; Bustillo, K. C.; Ercius, P.; Ciston, J.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Templated encapsulation of platinum-based catalysts promotes high-temperature stability to 1,100 degrees C. Nat. Mater. 2022, 21, 1290-1297.
23. Kim, Y. Y.; Keller, T. F.; Goncalves, T. J.; Abuin, M.; Runge, H.; Gelisio, L.; Carnis, J.; Vonk, V.; Plessow, P. N.; Vartaniants, I. A.; Stierle, A., Single alloy nanoparticle x-ray imaging during a catalytic reaction. Sci. Adv. 2021, 7, eabh0757.
24. Dolling, D. S.; Chen, J.; Schober, J. C.; Creutzburg, M.; Jeromin, A.; Vonk, V.; Sharapa, D. I.; Keller, T. F.; Plessow, P. N.; Noei, H.; Stierle, A., Probing Active Sites on Pd/Pt Alloy Nanoparticles by CO Adsorption. ACS Nano 2024, 18, 31098-31108.
25. Saini, S.; Halldin Stenlid, J.; Deo, S.; Plessow, P. N.; Abild-Pedersen, F., A First-Principles Approach to Modeling Surface Site Stabilities on Multimetallic Catalysts. ACS Catal. 2024, 874-885.