
Reaktionsmechanismen und Katalysatordesign
Das wesentliche Forschungsziel in der Gruppe „Katalysatoren von Kohlenwasserstoffen“ ist, Beiträge zur Entwicklung erneuerbarer Quellen von Kohlenstoff für den Energieverbrauch der Gesellschaft zu liefern. Durch Modellierung mit periodischer Dichtefunktionaltheorie (DFT) und ab initio Berechnungen an cluster Modellen erlangen wir ein Verständnis solcher Prozesse auf atomarer Ebene. Die wichtigsten Programmpakete, die wir verwenden, sind VASP, TURBOMOLE und ORCA. Die hauptsächlichen Themen in der Gruppe sind die Umsetzung von Methanol zu Kohlenwasserstoffen, die gezielte Funktionalisierung von Biomasse und die Untersuchung von Katalysatordeaktivierung.
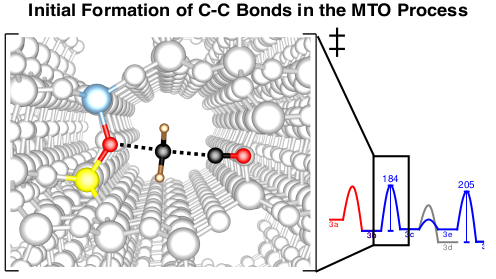
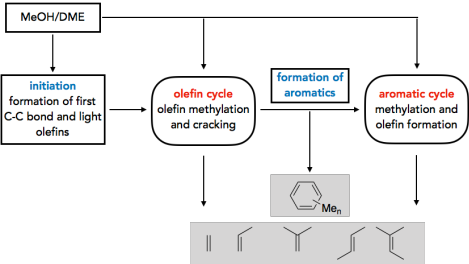
Synthese von Kohlenwasserstoffen aus Methanol
Es gibt ein großes Interesse an Prozessen, die Methanol selektiv in Produkte wie Kohlenwasserstoffe, Alkene und Kraftstoffe umwandeln. Das Verständnis der Reaktionsmechanismen kann hierbei zur Verbesserung von Selektivitäten und Ausbeuten der verwendeten Katalysatoren beitragen. Eine Frage von besonderer Bedeutung ist die Initiierung des methanol-to-olefins (MTO) Prozesses. Während es allgemein anerkannt ist, dass die Reaktivität im stationären Zustand durch katalytisch aktive Kohlenwasserstoffe bestimmt wird, die den sogenannten‚ hydrocarbon pool’ (HCP) bilden, ist weniger gut erforscht, wie sich dieser HCP bildet. Wir untersuchen dieses Problem durch die Berechnung vieler möglicher Reaktionen, wobei wir hochgenaue CCSD(T) und MP2-Rechnungen mit periodischen DFT-Rechnungen kombinieren1-3. Kinetische Modellierungen zeigen, dass die direkte C-C Bindungsbildung durch Methanol tatsächlich einen realistischen Mechanismus dargestellt, wobei der günstigste Mechanismus über die Bildung von CO und dessen weitere Methylierung abläuft.4 Neben der Initiierung untersuchen wir auch die Rolle der Olefine5 und Aromaten6 im HCP.
Methoden zur genauen Berechnung von Reaktionsmechanismen in Zeolithen
Hochgenaue ab initio Methoden, wie DLPNO-CCSD(T) in Kombination mit complete basis set (CBS) extrapolierten MP2 Rechnungen lassen sich nur für nichtperiodische cluster Modelle durchführen. In der Praxis lassen sie sich daher nur auf stationäre Punkte anwenden und werden mit der harmonischen Näherung für die Bewegung der Atomkerne kombiniert. Die Dynamik der Kerne lässt sich genauer und allgemeiner mit semiklassischen Born-Oppenheimer Molekulardynamik (MD) Rechnungen simulieren. Intrinsische Reaktionsbarrieren adsorbierter Spezies lassen sich mithilfe von Techniken wie umbrella sampling oder blue moon sampling berechnen. Ein zentrales Problem stellt hierbei die Adsorption und Desorption von Edukten und Produkten dar, die für eine genaue Beschreibung von Katalyseprozessen essentiell ist. Hierzu waren wir an der Entwicklung verschiedener Methoden beteiligt, mit denen sich freie Energien für Adsorption berechnen lassen12-14. Zusammen mit free energy perturbation theory (FEPT) lassen sich nun freie Energien mit MD Simulationen, mit denen Vorhersagen für Reaktionskinetiken auch für komplexe Reaktionsmechanismen mögliche sind15.
Struktur und Dynamik von Übergangsmetallkatalysatoren
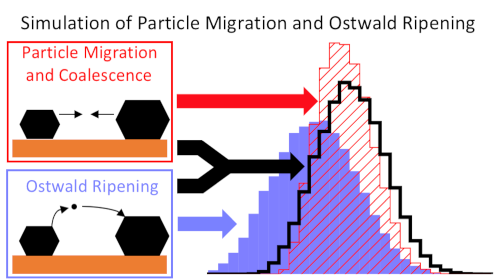
Theoretische Untersuchungen konzentrieren sich häufig auf die Berechnung von Reaktionsmechanismen anhand von Modellen der aktiven Zentren, wie der fcc(111) oder fcc(211) Oberfläche. Unser Ziel ist es, die Struktur und Stabilität der Katalysatoren unter Reaktionsbedingungen vorherzusagen. Neben den Oberflächenenergien ist die Wechselwirkung mit dem Trägermaterial (meist einem Oxid) der wichtigste Faktor für die Form der Metallpartikel. Die Untersuchung von Oberflächen und Grenzflächen unter Berücksichtigung der Reaktionsbedingungen mit DFT liefert deren Stabilität und erlaubt die Vorhersage der Gleichgewichtsstruktur16-18. Weiterhin untersuchen wir die Deaktivierung von Katalysatoren über Sinterung19-22. Neben monometallischen Katalysatoren untersuchen wir außerdem die Eigenschaften legierter Katalysatoren18, 23-25. Hierbei verwenden wir auch machine learning Modelle, die neben der Stabilität auch spektroskopische Eigenschaften von Adsorbaten vorhersagen können 26.
Publikationen
1. Plessow, P. N.; Studt, F., Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations. ACS Catal. 2017, 7, 7987-7994.
2. Goncalves, T. J.; Plessow, P. N.; Studt, F., On the Accuracy of Density Functional Theory in Zeolite Catalysis. ChemCatChem 2019, 11, 4368-4376.
3. Huber, P.; Plessow, P. N., The role of decarboxylation reactions during the initiation of the methanol-to-olefins process. J. Catal. 2023, 428.
4. Huber, P.; Plessow, P. N., Accurate and Efficient Description of Acidic Zeolites with Plane-Wave Density Functional Theory Using Range-Separated Hybrid Functionals. ChemPhysChem 2025, e2500147.
5. Plessow, P. N.; Smith, A.; Tischer, S.; Studt, F., Identification of the Reaction Sequence of the MTO Initiation Mechanism Using Ab Initio-Based Kinetics. J. Am. Chem. Soc. 2019, 141, 5908-5915.
6. Plessow, P. N.; Studt, F., Olefin methylation and cracking reactions in H-SSZ-13 investigated with ab initio and DFT calculations. Catal. Sci. Technol. 2018, 8, 4420-4429.
7. Fečík, M.; Plessow, P. N.; Studt, F., A Systematic Study of Methylation from Benzene to Hexamethylbenzene in H-SSZ-13 Using Density Functional Theory and Ab Initio Calculations. ACS Catal. 2020, 10, 8916-8925.
8. Plessow, P. N.; Enss, A. E.; Huber, P.; Studt, F., A new mechanistic proposal for the aromatic cycle of the MTO process based on a computational investigation for H-SSZ-13. Catal. Sci. Technol. 2022, 12, 3516-3523.
9. Plessow, P. N.; Studt, F., Cooperative Effects of Active Sites in the MTO Process: A Computational Study of the Aromatic Cycle in H-SSZ-13. ACS Catal. 2022, 13, 624-632.
10. Enss, A. E.; Plessow, P. N.; Studt, F., Theoretical investigation of the paring mechanism of the MTO process in different zeolites. J. Catal. 2024, 432.
11. Vicente, H.; Huber, P.; Gayubo, A. G.; Studt, F.; Plessow, P. N., Reactivity of Pentamethylcyclopentenyl Cations toward Olefin Formation in the Methanol-to-Olefin (MTO) Process. J. Phys. Chem. C 2025, 129, 20971-20980.
12. Amsler, J.; Plessow, P. N.; Studt, F.; Bucko, T., Anharmonic Correction to Adsorption Free Energy from DFT-Based MD Using Thermodynamic Integration. J. Chem. Theory Comput. 2021, 17, 1155-1169.
13. Amsler, J.; Plessow, P. N.; Studt, F.; Bucko, T., Anharmonic Correction to Free Energy Barriers from DFT-Based Molecular Dynamics Using Constrained Thermodynamic Integration. J. Chem. Theory Comput. 2023, 19, 2455-2468.
14. Plessow, P. N., Anharmonic Adsorption Free Energies and Apparent Activation Barriers for Mobile Reactants Based on Molecular Dynamics Simulations. J. Chem. Theory Comput. 2025.
15. Vicente, H.; Plessow, P. N., Formaldehyde formation and its impact on the MTO initiation investigated with ab initio molecular dynamics simulations. J. Catal. 2026, 457.
16. Chen, J.; Sharapa, D.; Plessow, P. N., Stability and formation of hydroxylated α−Al2O3(0001) surfaces at high temperatures. Phys. Rev. Res. 2022, 4, 013232.
17. Plessow, P. N.; Campbell, C. T., Influence of Adhesion on the Chemical Potential of Supported Nanoparticles as Modeled with Spherical Caps. ACS Catal. 2022, 12, 2302-2308.
18. Hejral, U.; Plessow, P. N.; Franz, D.; Shipilin, M.; Gutowski, O.; Rütt, U.; Noei, H.; Vonk, V.; Stierle, A., Composition-Dependent Alloy Nanoparticle Shape Changes under Reaction Conditions: Kinetic and Thermodynamic Effects. J. Phys. Chem. C 2024, 128, 4330-4342.
19. Plessow, P. N.; Abild-Pedersen, F., Sintering of Pt Nanoparticles via Volatile PtO2: Simulation and Comparison with Experiments. ACS Catal. 2016, 6, 7098-7108.
20. Goodman, E. D.; Johnston-Peck, A. C.; Dietze, E. M.; Wrasman, C. J.; Hoffman, A. S.; Abild-Pedersen, F.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Catalyst deactivation via decomposition into single atoms and the role of metal loading. Nat. Catal. 2019, 2, 748-755.
21. Goodman, E. D.; Carlson, E. Z.; Dietze, E. M.; Tahsini, N.; Johnson, A.; Aitbekova, A.; Nguyen Taylor, T.; Plessow, P. N.; Cargnello, M., Size-controlled nanocrystals reveal spatial dependence and severity of nanoparticle coalescence and Ostwald ripening in sintering phenomena. Nanoscale 2021, 13, 930-938.
22. Aitbekova, A.; Zhou, C.; Stone, M. L.; Lezama-Pacheco, J. S.; Yang, A. C.; Hoffman, A. S.; Goodman, E. D.; Huber, P.; Stebbins, J. F.; Bustillo, K. C.; Ercius, P.; Ciston, J.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Templated encapsulation of platinum-based catalysts promotes high-temperature stability to 1,100 degrees C. Nat. Mater. 2022, 21, 1290-1297.
23. Kim, Y. Y.; Keller, T. F.; Goncalves, T. J.; Abuin, M.; Runge, H.; Gelisio, L.; Carnis, J.; Vonk, V.; Plessow, P. N.; Vartaniants, I. A.; Stierle, A., Single alloy nanoparticle x-ray imaging during a catalytic reaction. Sci. Adv. 2021, 7, eabh0757.
24. Dolling, D. S.; Chen, J.; Schober, J. C.; Creutzburg, M.; Jeromin, A.; Vonk, V.; Sharapa, D. I.; Keller, T. F.; Plessow, P. N.; Noei, H.; Stierle, A., Probing Active Sites on Pd/Pt Alloy Nanoparticles by CO Adsorption. ACS Nano 2024, 18, 31098-31108.
25. Saini, S.; Halldin Stenlid, J.; Deo, S.; Plessow, P. N.; Abild-Pedersen, F., A First-Principles Approach to Modeling Surface Site Stabilities on Multimetallic Catalysts. ACS Catal. 2024, 874-885.
26. Chen, J.; Sharapa, D.; Plessow, P. N., Prediction of Vibrational Spectra of CO Adsorbed on PdPt(111) Using Machine Learning and Monte Carlo Simulations. ACS Catal. 2026, 16, 8784-8797.